Bei LMTO47 gibt es eine Vielzahl von physikalischen und numerischen
Optionen, die ebenfalls Einfluß auf das Ergebnis haben: ich habe
jeweils so viele Leerkugeln verwendet, daß der Kugelüberlapp nicht
über 25% stieg, und für die selbstkonsistenten Rechnungen die
Zahl der k-Punkte jeweils so lange erhöht, bis die DOS sich dem
Augenschein nach nicht mehr nennenswert änderte. Da aus magnetischen
und Mößbauer-Messungen bekannt ist, daß i-AlCuFe und
i-AlPdMn nicht ferromagnetisch sind, wurden in den Rechnungen (bis auf
elementares Eisen und Mangan) keine Spinorbitale verwendet. Gelöst
wurde eine skalare relativistische Wellengleichung, als Austausch- und
Korrelationsterm wurde das lokale Dichtefunktional von von Barth und
Hedin verwendet. Die Berechnungen wurden je nach Umfang auf PCs,
DEC-alpha Workstations oder einer IBM RS/6000 durchgeführt.
Zur Berechnung der Bindungsenergien wurden mit der Option FREE totale
Energien der freien Atome zu vorgegebener Elektronenkonfiguration
(siehe [Kittel]) bestimmt: Sie sind rechts in
Tabelle
D.1 zu finden.
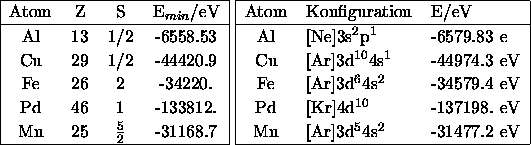
Tabelle D.1: Energien freier Atome, berechnet mit Gaussian94 und LMTO
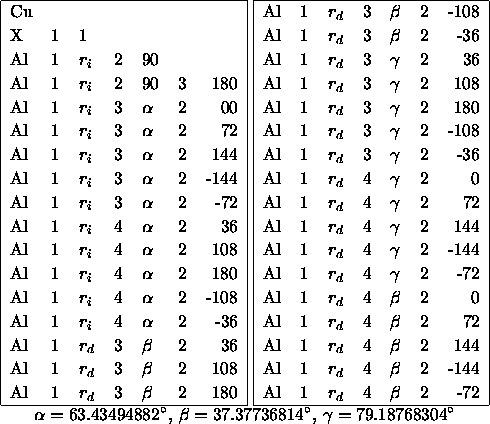
Tabelle D.2: Z-Matrix des Bergmanclusters